
All you need to know about the Usher Syndrome and Related Disorders Conference, Part III: Genes, Proteins, and Networks
September 15, 2010
by Jennifer Phillips, Ph.D.
We're slowly progressing through the wealth of interesting talks at the Conference last May, and even though I didn't actually attend the Valencia meeting, I have enough familiarity with the dense molecular topics therein to give an adequate summary and their relevance to the Usher community. My plan is to cover the Usher gene overviews along with a few studies of particular players in Part III, and recap the talks that dealt with the topics of new genes and related genetic disorders in Part IV.
Prologue: It should come as no surprise to the readers here that Usher syndrome has multiple genetic causes-that is, mutations in one of twelve (so far) distinct chromosomal regions known as loci result in deafblindness in humans. Even though there are important differences in the onset, and precise symptoms of Usher syndrome, there are obviously enough commonalities to treat all cases of Usher as one general disorder. As such, one of the main research goals for those of us who study these molecules is the two-pronged investigation of
a) what they have in common, i.e. why defects in all these different genes cause a very specific type of sensorineural deafness and retinal degeneration, and
b) what makes each gene different, i.e. why mutations in some genes cause more severe forms of Usher than others.
So with that in mind, let's get started on the summaries...
The Usher protein network in the inner ear
Hannie Kremer, Radboud University Nijmegen, The Netherlands
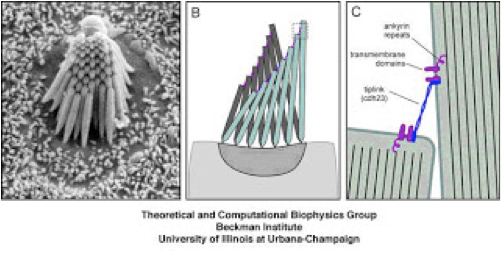
Figure 1: A-an electron microscope scan of the actin bundle, made up of organized stereocilia, projecting from a single hair cell. B-a cartoon of these structures showing a passive mode (gray, background) and an active mode (green, foreground), in which the stereocilia bend in unison in response to an incoming sound wave. C. A cartoon showing a magnified view of the tips of two individual stereocilia (green) tethered together by the Usher protein, Cadherin 23.
The above figure shows the organization of a specialized structure found in mechanosensory hair cells. The hair bundle is the structure that give 'hair cells' their name, although it's not really made of hair, but actually comprised of tubes of actin that protrude in the same direction from the tops of these cells and are connected at the tips so that they will move as a unit in response to vibration, causing the hair cell to signal the auditory neurons that a sound has been perceived. Mice with mutated or deleted Usher genes have clear and specific defects in the formation of stereocilia, the actin bundles that project from the tops of the mechanosensory hair cells:
Figure 2: from Lefevre et al, 2008. The far left panel shows several hair cells (outer hair cells (OHC) and inner hair cells (IHC) )from the cochlea of a mouse without an Usher mutation. The rest of the panels are images from this same region of the cochlea from mice with defects in Usher genes. From left to right: wild-type, Myo7a, Ush1c, Cdh23, Pcdh15, Ush1g.
The observed defects in the length, width, orientation and abundance of stereocilia in the Usher mice show a clear requirement for Usher proteins in the proper formation of these structures. As all of this growth and development takes place before birth in humans, it's no surprise that the hearing loss in most Usher cases is congenital. But what exactly makes the stereocilia so disorganized? We know from biochemical studies that many of the Usher proteins can physically interact with each other, and we know from antibody localization studies that many of the Usher proteins localize to particular regions of the hair bundles, so detailed studies of these in combination with the mouse Usher mutants provides evidence (alluded to in figure 1C) that the Usher proteins work together in a complex to guide the organized growth of the actin bundles by linking the individual stereocilia together. In addition to this proposed function of an Usher complex, individual Usher proteins appear to play a role in the outgrowth of the actin structures. Finally, there is some, less-well understood evidence that some Usher proteins may also be involved in events at the other end of the hair cell-the part that communicates with the auditory neurons.
Why you care
Understanding when and how the Usher proteins are required for normal cellular development and function is critical for the discovery of targeted therapies. The more we can find out about what these molecules are doing in there, the better chance we have of identifying ways to compensate for defective proteins in Usher patients.
The Usher proteins in the retina
Uwe Wolfrum, Johannes Gutenberg University of Mainz, Germany
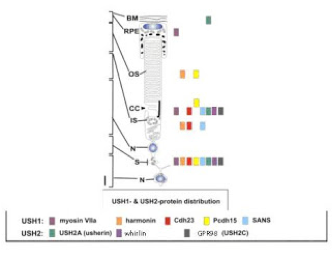
Figure 3: Usher protein localization in subcellular regions of the photoreceptor cell. Abbreviations from top to bottom: BM: basement membrane; RPE: retinal pigmented epithelial cell; OS: outer segment; CC: connecting cilium; IS: inner segment; N: nucleus; S: synapse; N: neuron.
adapted from Reiners et al, 2006.
One of the confounding things about Usher syndrome is the asynchronous timing of the hearing and vision defects. We have good evidence from the mouse models that the congenital hearing loss is due to developmental defects, but the vision loss is progressive, which would suggest a problem with function rather than with formation. In all the human and animal models we currently have access to, there doesn't appear to be anything wrong with the structure of the retina, but something is causing photoreceptors to die, slowly, over a course of months or years. Again, researchers can seek answers by looking at the protein localization within the retina, and from knowing that these proteins have the potential to physically interact with each other to form multi-molecular complexes. Because it's the photoreceptor cells that degenerate, most of the protein localization studies have focused on this cell type. Similar to the colocalization pattern seen in hair cells, many Usher proteins conglomerate in distinct functional regions of the photoreceptor cells. One exciting finding is that many of these proteins (indicated by colored boxes in figure 3) localize in the region of the connecting cilium. The connecting cilium in photoreceptors serves as a conduit for moving proteins and other molecular cargo that are synthesized in the cell body, or inner segment, up to the outer segment where they're required to perform various functions. The abundance of Usher proteins at this particular location has prompted researchers to examine whether any of them might interact with known factors in molecular trafficking, and indeed, some do. Although the evidence is still being gathered, it's tantalizing to imagine that defects in cargo loading or transport, or indeed, in the structure of the conduit itself, might be responsible for the reduced function and ultimate degeneration of photoreceptors. This is a very hot area of research and new ways of assessing these functions are being developed in numerous labs worldwide. Additionally, and not unlike the situation in hair cells, there is some evidence that these proteins localize to and function somehow in the synapse, through which the photoreceptor signals to adjacent neurons when stimulated.
Why you care
Again, understanding the where, when, and how of Usher protein activity is essential to finding therapeutic ways to overcome defects in these processes. These are incredibly complex cells, and there is strong evidence that Usher proteins perform multiple functions within them, so there is still a great deal to be learned, but every new bit of data adds to the big picture that will someday reveal new ways to address or prevent this progressive vision loss.
Usher scaffold proteins provide complementary functions in retina and inner ear
Monte Westerfield, University of Oregon, Eugene, Oregon, USA
Most of the Usher gene studies using animal models have been conducted in mice or rats, but a few of us are using the zebrafish to address these questions. Zebrafish have hair cells and retinas very similar those found in humans and these animals provide a great system in which to study the consequences of targeted Usher gene or protein disruption. In the above summaries, I alluded to the fact that Usher proteins display the ability to physically interact in a complex. This is achieved through the presence of particular protein interaction domains, some of which provide a place to stick, and others of which are capable of binding to that sticky place, rather like the hook and loop surfaces found in Velcro. Several Usher proteins contain multiple sticking places, called PDZ domains, along their length, and are thought to be required inside a particular cell (e.g. a photoreceptor or a hair cell) to serve as a scaffold or an organizing structure for localizing other Usher proteins that may function across cell boundaries, or, in the case of the links between stereocilia depicted in Figure 1C, way up the page, between membranes of structures from the same cell. Interestingly, although there are characteristics that make PDZ domains identifiable, there appears to be a great deal of specificity in which PDZ domains certain Usher proteins with PDZ-binding capabilities choose to interact with. In other words, even though there are a couple of different Usher proteins, namely Ush1c and Ush2d, that look very similar with respect to the presence and placement of PDZ domains, they don't appear to have very much functional overlap. Is this because they're not in the same place at the same time, or are there other regulatory factors that influence which of these proteins is the more suitable 'scaffold' in a given cellular situation? We are pursuing these questions, as well as ones of general function of all the involved PDZ proteins. Zebrafish in which these PDZ proteins have been disrupted appear to have similar functions in hair cells to what has been shown in mice, with respect to the development and organization of stereocilia. We can also detect functional problems at the behavioral and cellular level. In the retina, we see specific defects in synaptic transmission between photoreceptors and retinal neurons when we disable one of the PDZ domain containing proteins (Ush1c) and we've also discovered that Ush1c and Ush2d proteins are all present in another retinal cell type called the Müller glia. Müller glial cells have a variety of functions related to maintaining the health, development and function of retinal neurons, including photoreceptors, so the possibility that these cells could somehow be involved in the defects observed in Usher syndrome is intriguing.
Why you care
Investigating Usher gene function in organisms other than Mammals adds a wealth of additional experimental techniques with which to understand the role of Usher proteins at various developmental and functional time points in the ear and the retina. In addition to the general benefit of obtaining more information about the complex workings of these molecules, the zebrafish system also lends itself to high-throughput testing of various genetic and molecular therapies.






