
Gene Therapy for Retinal Disease: What Could It Mean for Usher Patients? Part I
September 25, 2009
By Jennifer Phillips, Ph.D.
As promised, I'm going to write about some good science news here. However, I must implore you not to forget everything I've told you about the pace of research. Cautious optimism is warranted, so feel free to do a little chair dance if you must, but please don't call your Ophthalmologists straightaway and ask them about this new cure you've just read about on the Usher Syndrome Blog. Although the research I'm going to describe below might, in time, get us closer to a cure, we're not quite there yet.
The research I want to share with you involves gene therapy for another retinal disease that shares some similarities with Usher syndrome. However, before I jump right into the research findings, I need to fill you in on some general information about gene therapy. My plan is to use this post to give you the background and describe the research, and then connect the dots to Usher syndrome in my next post.
So let's start with some basics: what is 'gene therapy', anyway? When we talk about gene therapy we're usually referring to 'gene replacement', whereby a healthy, normal copy of a gene is introduced into the body of an individual whose own copies of this gene are absent or abnormal in some way. There are several different delivery systems under study, but the ones that have been the most efficient overall have been modified viruses.
A virus, as you may recall from Biology 101, is unlike anything else that we consider strictly 'alive' in that it isn't composed of cells, and thus lacks the molecular machinery that allows every other form of life to survive, function, and proliferate. Instead, viruses require a host cell to do their replicating for them. Viruses are streamlined for this task, consisting of genetic material (in the form of DNA or RNA) wrapped up in a package of proteins that help it enter the cells of its target host. Once a virus has gained access to a cell, its genetic information can be copied and used to build functional proteins in the same ways that a cells makes functional proteins from the information encoded by their native genes. You can think of a virus as kind of a freeloading office worker who slips a few of his personal papers in with a large copy job for the company.
In using viruses for use in gene therapy, scientists have essentially turned the tables on the viruses. Viruses have exploited the cellular machinery of all living things on this planet for millions of years. Now, because the mechanisms of viral infection are so well understood, we can exploit what is useful about them, e.g. getting genetic material into cells, and inactivate or remove the other parts of viruses that aren't so wonderful, e.g. their tendency to replicate like crazy, stimulate your immune system, and make you sick.
During the modification process, the viral genes that promote the formation of new viruses and invasion of more host cell are removed and, replaced by the coding information for a complete, healthy gene of interest. Once this is complete, the next steps involve introducing it into the host cells. Once inside, the virus and its cargo will be replicated, serving as a blueprint for making biologically active levels of the protein that was previously missing or defective. Here's a diagram of this process:
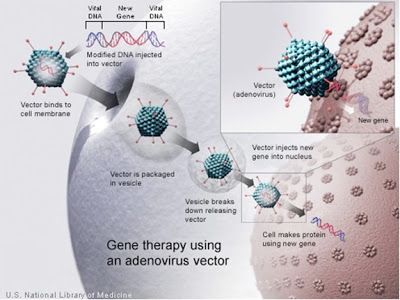
Figure 1: A modified adenovirus is used to deliver a new gene into a cell. The 'vector' is the virus, and the knobly exterior that resembles a ball of yarn with knitting needles sticking out of it represents the viral proteins surrounding the genetic material that enable the virus to enter the cell.
The particular kind of virus used in the above schematic, an 'adenovirus', may sound familiar to you because adenoviruses in their unmodified forms are the ones that cause the common cold, among other things. I hope it gives you some degree of satisfaction to know that their hitherto irritating power to infect is even now being harnessed to benefit humankind.
The next detail to deal with is the matter of getting the virus, and therefore the gene, into the right tissue. The disease being treated will usually dictate where the replacement gene is needed. If the symptoms are particular only to the brain, or the bone marrow, etc. efforts are made to deliver the gene to those specific locations. This often requires getting the viruses to infect cells in a petri dish in the lab and then introducing the cultured cells into the appropriate body tissue. For retinal delivery, however, researchers have found that directly injecting a small volume of liquid containing many copies of the viral vector into the back of the eye is effective in allowing the viral vectors to reach their target host cells in the retina.
So, with all those details in mind, on to the results:
Last year, two groups of researchers who have been working for some time on gene therapy for a type of Leber's congenital amaurosis (LCA) reported some results from their first clinical trials. Like Usher patients, individuals with LCA suffer from progressive vision loss beginning in childhood. Although the specific molecular reasons for this are quite different from what happens in Usher syndrome, the end result is the same-diminishing visual response over time, and a gradual thinning of the retina due to the death of photoreceptors. Importantly, the visual abnormalities (e.g. diminishing ERG response) in at least some types of LCA precede the cell degeneration. That is, the defective gene is present from birth, but it takes a long time-years, even decades-for the photoreceptors to actually die from the lack of a functional LCA protein. Again, this is very similar to what we see in at least some types of Usher syndrome. Yet another similarity between LCA and Usher syndrome is that a number of different genes can be responsible for the disease.
These researchers gave a lot of thought to which of the known LCA genes they would use in their study, selected a good candidate (RPE65, the gene implicated in LCA type 2), and tested it out in animal models-first rodents, and later dogs, all of which had mutations in this same LCA gene and as a result had a similar retinal defect to human LCA patients prior to their treatments. This part of the study took years, not only to perfect the delivery technique, but to monitor safety, determine effective doses, and evaluate short and long term changes in the vision of these test animals. At the end of this pre-clinical testing period, the animals were doing well, suffering no ill effects from the treatment, and showing improvement in their symptoms. Such positive results, at least in terms of safety, are required before moving on to human subjects. In this case, the pre-clinical results were very strong, and approval was given to begin Phase I Clinical Trials.
The number of subjects in a Phase I trial is usually very small: in each of these parallel LCA Phase I trials, three patients were analyzed. The subjects of these experiments were young people between the ages of 17 and 26. All had experienced vision loss, and all had some thinning of their retinas due to cell degeneration. However, they were all young enough that a good number of retinal cells remained, even though many of those cells were already functioning poorly and contributing to their visual symptoms.
All subjects underwent the ophthalmic surgery (under general anesthesia) to inject the viral vector into the space behind the retina of one eye. The other eye was left untreated so that 'before and after' type comparisons could be made in the same patient. The patients were monitored in the days and weeks following the surgery to make sure than none suffered any adverse effects from the surgery itself or the fact that a modified virus had been introduced into their bodies. All did well during and after the procedure, and beginning about one month after the treatment, one patient from one study and all three patients from the other began to exhibit improved visual function. These patients showed significantly increased responses to light in the injected eyes, and they experienced expansions in their visual fields. Both of these responses suggest that the presence of a healthy copy of the LCA2 gene was able to restore some of the function of the surviving retinal cells. Also encouraging is the fact that these effects seem to be
Phase I trials such as this are primarily designed to test safety and effective doses in humans, hence the small number of subjects to start with. You don't want to give a drug or treatment to a large group of people until you have a pretty good idea of what it's going to do to them. However, if everything goes well, safety-wise, in a Phase I trial, some data can be obtained on the effectiveness of the treatment as well. Happily, this was the case with these LCA trials, and even though the sample size is small and the data are still preliminary, the results were promising enough jto warrant publication. Here's a short (~10 minute) video of one of the researchers, Dr. Robin Ali, describing the trial:
So, to sum up the results of the studies: A gene that is defective in a progressive retinal degenerative disease can be safely introduced via viral vector into the retina, and can improve visual function for a sustained period of time. Several more phases of clinical trials must occur before this particular gene therapy can be offered in a clinical setting to LCA patients, but the promise of a cure is exciting indeed.
References:
Bainbridge et al., (2008) Effect of gene therapy on visual function in Leber's congenital amaurosisThe New England Journal of Medicine358 (21):2231-2239.
McGuire et al., (2008) Safety and efficacy of gene transfer for Leber's congenital amaurosis. The New England Journal of Medicine 358 (21):2240-2248.
Cideciyan et al., (2009) Vision 1 year after gene therapy for Leber's congenital amaurosis. The New England Journal of Medicine361:725-727.






