
New Usher Research in 2013
March 5, 2013
by Jennifer Phillips, Ph.D.
A couple of new stories/updates that will probably be of interest to our readers:
Thing 1: If you’ve followed my daily blogs from the ARVO meeting each May, and/or if you dialed into hear the November Conference Call, you should recall hearing about the potential treatment for USH1C that Jennifer Lentz and colleagues are testing in a mouse model. Details on the effects of this treatment on hearing and balance defects in the mouse were recently published in the journal Nature Medicine .
To recap the major results, this is a potential treatment being developed for a specific mutation in the gene that’s common in the Louisiana population of USH1C patients. I’ve written about this Ush1c mouse model before but here’s a primer on the nature of the mutation and how it lends itself to this type of therapy to help clarify things here:
A gene, as you may know, is a discrete package of information encoded by the base sequence of DNA. Individual chromosomes, long strands of DNA encoding many hundreds or even thousands of genes, reside permanently in the nuclei of our cells. The gene is the recipe for making a functional molecule—usually a protein—that, once made, will go out into the cell and do something. As part of the conversion process of taking the static information in the recipe and making it into an active protein, a molecule called RNA is used as a messenger. RNA is a similar molecule to DNA, and represents a faithful copy of the genetic information needed to make Protein A from gene A.
However, the message must undergo a bit of editing before it’s ready to serve as a template for building a protein. The simple figure below illustrates some of the steps:
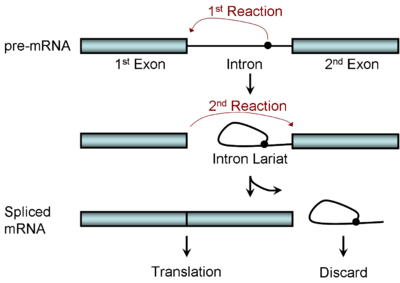
Figure 1. RNA splicing: a stepwise editing process. Image from http://en.wikipedia.org/wiki/RNA_splicing
In this figure, the ‘pre mRNA’ molecule at the top is an exact copy of the genetic information on the chromosome, but it contains extra information that needs to be edited out. Other molecules function as editors, locating the regions of the genetic code that need to be excised, cutting out the extra information, and splicing the remaining ends together to create the finished protein recipe that reads through. This process is kind of like what an extremely literal person might do if he or she found a great recipe published in a magazine, but the recipe was interrupted every few pages by a bunch of ads for Neutrogena products. The logical thing for that literal person to do would be to remove the ad pages so that the recipe would read straight through, and Neutrogena anti-aging skin cream wouldn’t accidentally be added to the Chicken Parmesan.
The molecules that convert (or ‘translate’) the information from the messenger RNA molecule into the building blocks of a protein fit the definition of extremely literal, so the message provided must be complete, correct, and contain no extraneous information. The figure above depicts the identification and removal of the extra bit labeled ‘intron’, which is then discarded as the remaining blue bars (the exons) are connected into one continuous piece of code. How do the molecules that perform this editing know where to make the cut? The instructions for how and where to do that are also in the DNA code—it contains information for its own editing.
When we talk about mutations, we are referring to changes to the DNA code that somehow affect the way the protein (or other functional molecules) is constructed. The message is faithfully copied, and that includes any mistakes that are in the original. Many mutations affect the part of the code that specifies which protein building blocks to include. Sometimes a mutation can change the code to specify a different building block, effectively swapping one ‘ingredient’ for another, which can affect the structure and function of the resulting protein. Other mutations will change the code such that information that used to specify a particular building block now codes for the end of the protein, like if a recipe was cut off too early after only the first four ingredients were listed.
Still other types of mutations affect the parts of the DNA that indicate where the editing, the cutting and splicing, should happen, so instead of extracting only the Neutrogena ads, parts of the actual recipe are accidently cut out as well. This third type is the kind of mutation is the one that’s common to the Acadian population in Louisiana and Canada.
In these patients, the gene that encodes information on making the USH1C protein contains a mistake that indicates a new, incorrect splice site. When this mistake gets incorporated into the pre-mRNA molecule depicted at the top of Figure 1, an important piece of the code is spliced out, and all the coding sequence from that point on is misread, resulting in a non-functional protein.
This mutation is called “216G>A” , which is shorthand for “the base at position 216 is mutated from a G to and A”, and that one small change specifies a splice site where there shouldn’t be one. The 216G>A mutation was identified by analyzing the DNA and messenger RNA of USH1C patients, but to study the exact effects of the change on developing or functional eye and ear tissue, an animal model was needed.
The news story linked to in the first paragraph describes a mouse model that was developed by researchers in Louisiana, which contained the same 216G>A mutation found in human patients. Once this mutant line was established, the researchers discovered that it exhibited all the symptoms of Usher type 1C: severe hearing problems, balance defects, and progressive vision loss. thus providing Lentz and colleagues with the opportunity to test therapies on the full range of symptoms.
Due to all the past research of the nature of this mutation, Lentz and colleagues have been able to focus their efforts on developing a therapy that will specifically combat incorrect splicing at this specific spot. They achieved this by creating a small molecule that sticks to the pre mRNA strand right where the mutated information is located. This small molecule then physically blocks the activity of the team of cutting and splicing molecules that come by, so that only the incorrect splice site is skipped over, while normal cutting and pasting occurs at all points before and after the mutated site.
With this recent work, Lentz and colleagues have now shown that using this small molecule reduces the presence of the incorrect message significantly. While some of the message molecules produced are still processed incorrectly, normally spliced mRNA is also present, thus creating a complete template for making the USH1C protein.
When Lentz and colleagues injected this molecule into baby 216G>A mice to see how this partial repair of the message might affect Usher symptoms, they observed that the circling behavior indicative of balance problems was absent, compared to 216G>A littermates who received control injections. They also noted significant improvements in performance on hearing tests, and an improvement in the condition of the cells within the inner ear that respond to sound waves. Some 216G>A mice received this treatment as early as three days after birth, while others received a single treatment at later time points: several days or up to two weeks after birth. Only when the treatment was given at 16days after birth did they see a reduction in the rescue effect. Thus, they were able to identify a fairly broad window in which treatment was effective. Encouragingly, even with one single treatment in these early days, the mice retained their hearing improvements up until about 6 months of age, when it started to decline again, and the improvements to balance were apparently retained at all stages studied so far. More encouraging still, the researchers knew that this wasn’t a complete rescue. Some of the ‘how to build Ush1c’ message molecules were still coming out wrong, but even a partial restoration of the normal message was enough to alleviate the hearing and balance symptoms significantly.
It’s important to point out here, again, that the hearing development in mice follows quite a different timeline than it does in humans. Baby mice are born deaf and don’t really start to hear until about the 6th day after they are born. Their auditory system continues to develop for several days after that, and in fact the point at which it can be considered ‘finished’ is about the point where the treatment failed to produce a result—16 days after birth. Human ears go through a similar developmental period, but it all takes place during fetal development, long before birth. Thus, access to this treatment would be somewhat more of a challenge were it to be considered for human use.
It remains to be seen what effect this molecule might have on the vision of these mutant mice, but the degree of hearing and balance rescue seen after a single dose is truly impressive. We’ll be following this story with great interest as it progresses.
Thing 2: I’ve written previously about the gene therapy trials for Leber’s Congenital Amaurosis, an early onset inherited form of retinal degeneration. The big news a few years ago was that gene therapy in Phase I trials, testing for safety and correct dosage, led to remarkable improvements in vision for the patients in the study. Prior to this clinical testing, they’d used the treatment on a canine model* of LCA type 2, with mutations in the same gene, RPE65, that is defective in humans suffering from LCA type 2. The original finding was that when RPE65 dogs were given the treatment early on, it improved their vision and reduced the degree of retinal degeneration that normally results from the disease—in dogs as well as humans.
The first reports of vision improvement in the human study participants have been followed up with more trials and observation of the subjects for longer periods of time. The good news is that all subsequent studies have consistently reported improvement in vision and/or retention of usable vision above what is normally seen with this progressive disease.
In a recent article published in the journal Proceedings of the National Academy of Sciences, researchers report that while the treatment shows long-term visual improvement, it apparently does nothing to diminish the retinal degeneration in these patients. Although the viable photoreceptors in treated eyes seem to be functioning better, they are still dying off at the slow, progressive rate seen in untreated eyes. Interestingly, they went back to the dog model to confirm this finding. All previous preclinical testing of this treatment, which showed improvement of vision AND reduction of retinal cell death, had been done on young dogs with the disease. When RPE65 dogs with blindness comparable to that of the human subjects who participated in the clinical trials were selected for treatment, researchers saw the same phenomenon in the dogs than they did in the humans: Improved vision, but NO delay in retinal degeneration.
Although this might seem a somewhat discouraging development, it is undoubtedly a good thing to know. If repairing cell function alone isn’t sufficient to prevent eventual cell death, we now know to look for treatments to improve cell survival rates that could be used in conjunction with the already established gene replacement therapy. We also know to watch for this phenomenon in future trials to test treatments for other forms of progressive blindness, including Usher syndrome. Meanwhile, it’s important to recognize that this is still a very effective treatment, even though it’s far from a perfect cure. Slowing the progression of vision loss is obviously not the same as stopping it, but it’s certainly better than nothing. Years of usable vision could be gained—years in which those eyes could see weddings, births, graduations, so much more; years in which an even better treatment might be discovered—before advancing degeneration ultimately outpaces improved cell function.
There’s a great deal to feel optimistic about in these studies, and many more are underway. Watch this space for further updates as they become available.
*to further the ongoing animal research discussion here, it seems important to clarify that researchers didn’t deliberately generate this mutation. Generations of selective breeding to propagate certain traits (coat color, ear shape, etc.) have resulted in some disease genes becoming more prevalent in certain dog breeds. Briards happen to have a mutation in RPE65 which, once identified, turned out to be useful for studying the human disease caused by the same gene.
References:
Lentz et al., 2013. Hearing and vestibular function by antisense oligonucleotide in a mouse model of human deafness.
http://www.ncbi.nlm.nih.gov/pubmed/23380860
Cideciyan et al., 2013. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement.






